摘要:心肌缺血导致心肌细胞死亡及心功能损害。现阶段,再灌注疗法被视为治疗心肌缺血的关键措施之一,然而其引发的心肌损伤问题亦不容忽视。在心肌缺血灌注再损伤(MIRI)的病理生理过程中,铁死亡作为一种细胞死亡形式,与氧化应激过程紧密相连,发挥着关键作用。抑制铁死亡从而减轻心肌细胞死亡,可能成为MIRI的有效治疗策略。活性氧(ROS)是铁死亡发生和发展的关键组成部分,因此研究ROS在心肌损伤细胞中铁死亡的机制,进而开发更有效和精准的缺血再灌注治疗方法,对于降低心血管疾病的社会和经济成本具有重要意义。
关键词:活性氧,铁死亡,心肌缺血灌注再损伤
1心肌缺血灌注再损伤的概述
心肌缺血是指心脏的血液灌注减少,导致心脏的供氧减少,心肌能量代谢不正常,不能支持心脏正常工作的一种病理状态。心肌缺血是冠状动脉粥样硬化的主要危害[1]。严重和持续的心肌缺血引起不可逆损伤即心肌梗死。目前临床医生认为通过再灌注恢复血流以挽救缺氧缺血组织是心肌梗死最有效的治疗策略[2],再灌注虽能挽救缺血的心肌但同时也可诱发不可逆损伤[3]。缺血引起的心肌细胞死亡、能量代谢紊乱、心功能受损等一系列损伤性变化,再灌注后表现得更为突出,甚至引发严重的心律失常导致猝死。这是心肌梗死死亡率增加和预后不良的原因,从而在很大程度上降低了再灌注治疗的优势。氧化应激是心肌缺血灌注再损伤(MIRI)发生的主要原因之一,活性氧(ROS)累积对心肌细胞和血管组织造成严重损伤。过量的ROS导致膜脂过氧化,破坏细胞膜的屏障功能,加重心肌细胞损伤并最终导致心肌细胞死亡,ROS的累积对心肌细胞的损伤严重且不可逆。当前面对MIRI这一复杂且棘手的难题,尚未发现有效的治疗手段。在临床实践中,常用的心脏保护干预手段,例如治疗性低温,确实能在一定程度上减轻再灌注损伤的影响。然而,关于这些干预措施是否能在心脏再灌注的关键时刻产生足够的刺激强度信号,进而在缺血期间发挥保护作用,目前仍存在不确定性。因此,开发更加有效的预防和治疗方法具有重要的临床意义。
2铁死亡的概述
铁死亡是一种依赖细胞内铁的累积引起毒性脂质过氧化物ROS升高的非凋亡细胞死亡形式[4]。当细胞内铁稳态被破坏时,过量的铁通过芬顿反应将过氧化氢和脂质过氧化物转化为ROS,ROS的积累会破坏细胞膜完整性[5-6],促进细胞死亡。Baba等[7]的研究证实铁死亡与心肌细胞死亡密切相关。在成年小鼠心脏缺血/再灌注时,铁在缺血心肌周围的心肌细胞中积累,过量的铁会导致心肌细胞死亡,而抑制ROS的产生会减轻心肌细胞的死亡。铁死亡抑制剂雷帕霉素靶蛋白(mTOR)机制靶点的发现具有重要临床意义,其可能通过控制心肌细胞铁代谢来影响铁死亡,保护心肌细胞免受铁死亡。Chen等[8]在小鼠心肌IRI模型中发现细胞铁水平升高,GPX4活性以及重组蛋白(FTH1)和GSH水平降低。研究证实心肌细胞中铁死亡与氧化应激密切相关,抑制铁死亡从而减少心肌细胞死亡很可能成为MIRI的有效治疗策略。因此,深入了解MIRI中细胞死亡机制,探究铁死亡过程,进而开发更有效和精准的缺血再灌注治疗方法,降低心血管疾病的社会和经济成本具有重要意义。
3 ROS和铁死亡的关系及其作用机制
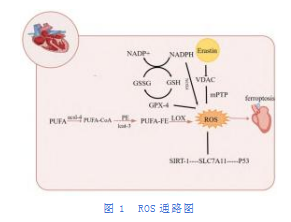
铁离子的过量积累可通过芬顿反应增加ROS的产生,影响铁的稳定性,促进铁在重要器官的沉积,从而导致严重的器官损伤。脂质ROS的形成是铁死亡发生和发展的关键组成部分。通过研究ROS在铁死亡中的作用机制(如图1),有利于分析和寻找靶点以减少心肌细胞铁死亡,通过分析整理归纳作用机制有以下几点。
(1)基于心肌缺血灌注再损伤中体内和体外模型的显示,系统Xc-是摄取胱氨酸的反向转运蛋白,由跨膜蛋白转运蛋白溶质载体家族7成员11(SLC7A11)和单道跨膜调节蛋白溶质载体家族3成员2(SLC3A2)组成,通过以1:1的比例交换谷氨酸和胱氨酸来调节铁死亡。通过SLC7A11起清除活性部位作用,ROS水平在MIRI心肌组织中显著升高,而SIRTUIN-1(SIRT-1)和SLC7A11表达下调,p53高度表达。过表达SIRT-1后,ROS水平降低,SLC7A11蛋白表达上调,p53蛋白下调。在S.Ma等[9]的实验中,进一步发现SIRT1通过p53/SLC7A11轴抑制铁死亡诱导的心肌细胞死亡。这表明ROS在铁死亡中起重要作用,调控ROS的表达,可能与SIRT-1/p53/SLC7A11信号通路有关。
(2)GPX-4作为内源性抗氧化剂,通过清除过量的过氧化物来保护细胞,从而减轻铁介导的脂质过氧化。GPX-4失活会诱导ROS升高,加重心肌细胞脂质过氧化,从而导致铁死亡。谷胱甘肽作为GPX-4的协同分子,辅助GPX-4分解过氧化氢,清除脂质过氧化物以保护细胞。抑制细胞内GSH和GPX-4的活性,可导致胞内ROS水平增加并最终介导铁死亡。在Feng等[10]的研究中表明,铁死亡抑制剂liproxstatin-1(Lip-1)可通过增加GPX4水平来减少积聚产生的ROS脂质过氧化,从而抑制铁死亡。在小鼠心肌缺血后给予Lip-1可减小心肌梗死面积,保护其心肌线粒体结构的完整性。
(3)ROS的产生依赖于多不饱和脂肪酸磷脂乙醇胺(PUFA-PE)。多不饱和脂肪酸(PUFA)能在酰基辅酶a合成酶长链家族成员-4(ACSL-4)的催化下酰化生成PUFA酰基辅酶a(PUFA-CoA),继而在溶血磷脂酰胆碱酰基转移酶-3(lcat-3)的作用下与磷脂酰乙醇胺(PE)反应生成PUFA-PE[11]。在脂氧合酶(LOX)的酶催化作用下,PUFA-PE对ROS的形成至关重要。在这一途径中,PUFA-PE的作用依赖于两个关键调节点,ACSL-4和脂氧合酶。多不饱和脂肪酸(PUFAs)被催化产生破坏性的脂质过氧化物,破坏细胞形态,如线粒体膜收缩和膜缺陷,最终导致铁死亡。因此,降低ACSL-4和脂氧合酶可以有效抑制PUFA-PE和抑制铁死亡的发生。
(4)ROS作为诱导因子介导线粒体自噬,调节心肌细胞铁死亡参与MIRI的发病机制。细胞自噬是一种通过溶酶体清除受损细胞器和蛋白质的精确调控的动态发展过程。铁死亡在一定程度上依赖于自噬,ROS可以通过氧化降低自噬相关蛋白的活性,从而引起自噬。VDAC是位于线粒体外膜上的传输离子和代谢物的通道蛋白,控制线粒体和其他细胞器代谢产物的交换。Erastin是典型的铁死亡诱导剂,在微管蛋白存在的情况下激活VDAC,引起线粒体超极化,线粒体通透性过渡孔(mPTP)的打开可导致线粒体ROS生成升高、膜电位损失和ATP耗竭,通过程序性或非程序性死亡机制诱导细胞铁死亡[19],造成心肌的再次损伤。
(5)NADPH氧化酶4(NOX4)是心肌细胞氧化应激的主要分子介质,主要功能是催化活性氧(ROS)的形成。NOX4将电子从NADPH转移到氧原子,甚至产生超氧化物。Doll S[12]发现,在心力衰竭模型中,敲低NOX4可通过改善铁超载而显著逆转心室重构,进一步推断药物抑制或敲除GPX4可有效预防铁死亡。ROS的过量产生引发的心肌细胞损伤与NOX4的升高具有一定的关联性。NAD(P)H氧化酶(Nox)作为心血管系统介导活性氧族生成的主要酶系之一,其介导的氧化应激对心脏缺血再灌注的影响及各亚型参与的应激机制的研究还需进一步深入。
4结语
众多研究发现,减少心肌细胞铁死亡可缓解心肌缺血灌注再损伤的发生,从而减少心肌细胞的损伤程度和面积。减少细胞铁死亡可能是未来提供心肌细胞损伤的潜在治疗方法的靶点之一。有研究显示,一种超短效、非巴比妥类催眠静脉麻醉剂依托咪酯通过Nrf2抑制IR诱导的铁死亡减轻了心肌损伤。但就目前的研究而言,对心肌细胞铁死亡、MIRI和心肌损伤的研究还存在一定的局限和缺憾。MIRI主要发生铁死亡的确切阶段尚未确定,一些研究表明,在MIRI的不同阶段,铁死亡的发生率不同,这就导致临床医生对患者用药和治疗的难度增加。因此,探索心肌细胞铁死亡的通路及机制与MIRI的联系还有很长的路要走。
参考文献
[1]Wang Y,Xue Y,Zhang T,et al.,2021.Photosynthetic biomaterials:applications of photosynthesis in algae as oxygenerator in biomedical therapies.Bio-des Manuf 4(3):596–611.
[2]Borja I,Stefan J,Stefan A,et al.2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation[J].European Heart Journal,2017(2):2.
[3]Ov ize,Michel,Garcia-Dorado,et al.Targeting r ep e rf us io n i nj u ry i n patie nts w ith ST-segment elevation myocardial infarction:trials and tribulations[J].European Heart Journal the Journal of the European Society of Cardiology,2017,38:935-941.
[4]Dixon S J,Winter G E,Musavi L S,et al.Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death.[J].ACS Chemical Biology,2015,10(7):1604-1609.
[5]Dixon S J,Lemberg K M,Lamprecht M R,et al.Ferroptosis:An iron-dependent form of nonapoptotic cell death[J].Cell,2012(5):149.
[6]Yangmin Qiu,Yue Cao,Wangjia Cao,et al.The Application of Ferro pto s is in Diseases[J].Pharmacol Res,2020 Sep:159:104919.
[7]Baba Y,Higa JK,Shimada BK,et al.Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes[J].Am J Physiol Heart Circ Physiol,2018(314):H659-68.
[8]Chen H Y,Xiao Z Z,Ling X,et al.ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating auto phagy[J].Molecular Medicine,2021,27(1):14.
[9]Shuxian Ma,Linyan Sun,Wenhao Wu,et al.USP22 Protects Against Myocardial Ischemia-Reperfusion Injury via the SIRT1-p53/SLC7A11-Dependent Inhibition of Ferroptosis-Induced Cardiomyocyte Death[J].Front Physiol,2020 Oct 21:11:551318.
[10]Feng Y,Mad un gwe N B,Aliaga n A D I,et a l.Lip rox statin-1 p rotects the mo use myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels[J].Biochemical and Biophysical Research Communications,2019,520(3):606-611.
[11]Brent R Stockwell,JoséPedro Friedmann Angeli,Hülya Bayir,et al.Ferroptosis:A Regulated Cell Death Nexus Linking Metabolism,Redox Biology,and Disease[J].Cell,2017 Oct 5;171(2):273-285.
[12]Doll S,Proneth B,Tyurina Y Y,et al.ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition[J].Nature Research,2017(1):91-98.
文章出自SCI论文网转载请注明出处:https://www.lunwensci.com/yixuelunwen/80339.html