SCI论文(www.lunwensci.com):
摘要:炎症小体在炎症性疾病发生发展中发挥了重要作用,其中 NOD 样受体 NLRP3( NACHT、LRR and PYD domains-containing protein 3, NLRP3) 炎症小体被机体各种内外源性危险信号激活后,通过活化半胱天冬酶-1(Caspase-1),进而促进 IL-1β、IL-18 的成熟和释放,引起机体的炎症反应,参与肥胖、2 型糖尿病、痛风、动脉粥样硬化等炎症性疾病的发生、发展。通过对 NLRP3 炎症小体的进一步研究将会加深人们对 NLRP3 炎症小体相关的炎症性疾病发生、发展的认识,从而更科学的掌握相关疾病的发病机制。本文将对近几年关于 NLRP3 炎症小体的激活模式、负调控及其与心血管疾病的关系的最新研究进展做一综述。
关键词:NLRP3 炎症小体;IL-1β;Caspase-1;心血管疾病
本文引用格式:刘旦旦 , 周静 , 高峰 . NLRP3 炎症小体及其与心血管疾病的关系 [J]. 世界最新医学信息文摘,2018,18(78):88-89,91.
New Development of NLRP3 Inflammasome and the Connection with Cardiovascular Diseases
LIU Dan-dan, ZHOU Jing, GAO Feng*
(Dongguan branch, Affiliated Hospital of Yan’an University, Yanan, Shaanxi, Yanan Shaanxi)
ABSTRACT: Inflammasome plays an important role in the development of inflammatory diseases, in which the NOD-like receptor NLRP3 inflammasome is activated by various endogenous and exogenous danger signals of the body and activates caspase-1 (Caspase-1). It promotes the maturation and release of IL-1β and IL-18, induces inflammatory reactions in the body, and participates in the occurrence and development of inflammatory diseases such as obesity, type 2 diabetes, gout, and atherosclerosis. Further research on the NLRP3 inflammasome will deepen people’s understanding of the occurrence and development of NLRP3 inflammatory body-related inflammatory diseases, so as to more scientifically grasp the pathogenesis of related diseases. This article will review recent advances in the study of the activation pattern, negative regulation of NLRP3 inflammasome, and its relationship with circulatory diseases.
KEY WORDS: NLRP3 inflammasome; IL-1β; Caspase-1; Cardiovascular diseases
0引言
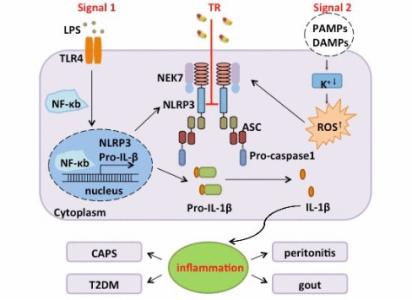
固有免疫,通过一类模式识别受体(Pattern recognition receptor,PRR)去识别病原生物表达的称为病原体相关模式分子(Pathogen-associated molecular patterns,PAMP)的结构, 进而诱导机体产生有效的适应性免疫应答防御病原体的入侵。NOD 样受体(Nod-like receptor)NLRP3 炎症小体,是目前研究最为火热的一种蛋白复合物,被激活后可作为分子信号平台,激活 caspase-1,调节强有力的促炎症细胞因子 IL-1β、IL-18 的成熟 [1]。最新研究表明,NLRP3 炎症小体的激活与高血压、动脉粥样硬化、心肌梗死等循环系统疾病的发生、发展有关 [2-3]。在这篇综述中,我们重点讨论 NLRP3 炎症小体的结构、激活机制、负调控,及其在循环系统疾病的关系。
1 NLRP3 炎症小体结构和功能
NLRP3 炎症小体是由 NOD 样受体蛋白 3(NLRP3)、凋亡相关斑点样蛋白 (ASC) 以及半胱氨酸天冬氨酸特异性蛋白酶 -1(Caspase-1) 组成的高分子量蛋白复合物 [4]。广泛存在于人体单核巨噬细胞、B 细胞、T 细胞等免疫细胞及非免疫细胞 [5]。NLRP3 炎症小体活化后,通过活化的 Caspase-1 持续地将 pro-IL-1β 和 pro-IL-18 剪切为成熟的 IL-1β 和 IL-18, 激活相对应的信号转导通路,释放大量炎性物质,参与糖尿病、高血压、动脉粥样硬化等炎症性疾病的发生发展 [6]。
2 NLRP3 炎症小体激活机制
NLRP3 炎症小体可被人体各种内、外源性信号激活 [7], 通过间接的方式来参与 NLRP3 炎症小体的活化,但其具体的激活机制尚不明确,目前较为认可的主要有以下三种:
(1)离子浓度变化:Petrilli 等发现人为的提高培养基中 K+ 浓度后,细胞内 K+ 外流及 NLRP3 炎症小体的活化均受到明显抑制 [8-9],有学者提出半通道 pannexin-1 及胞外配体(如 LPS)在其中起到了关键作用 [10]。此外,人为提高培养基中 Ca+2 浓度可诱导 NLRP3 炎症小体的活化 [11],其主要通过触发 G 蛋白偶联受体来激活炎症细胞 [12]。
(2)溶酶体破坏:巨噬细胞吞噬 NLRP3 炎症小体活化剂后,引起吞噬小体不稳定,导致溶酶体破碎,溶酶体组织蛋白 B(cathepsin B)等被释放到胞质中,激活 NLRP3 炎症小体。使用组织蛋白酶抑制剂可以阻断溶酶体破坏导致的 NLRP3 炎症小体活化 [13]。
(3)线粒体氧化:NLRP3 炎症小体活化剂使线粒体功能紊乱,生成大量ROS,促进 NLRP3 炎症小体的组装活化。Zhou 等向细胞内添加 ROS 抑制剂(乙酰半胱氨酸)后,可见胞内的 caspase-1 活化水平和成熟 IL-1β 的生成受到明显抑制 [14]。Shenker 等发现敲除线粒体外膜上的电压依赖门控通道后,ROS 的产生明显减少,NLRP3 炎症小体的活化明显被抑制 [15]。
3 NLRP3 炎症小体负调控
NLRP3 炎症小体过度活化会导致组织损伤,自噬通过清除炎症小体、细胞因子等来实现其对 NLRP3 炎症小体负调控。自噬相关蛋白 16L1(Atg16L1)缺失将促进内毒素诱导炎症的发生 [16]。自噬相关蛋白 1 轻链 -3B(LC3B)的缺失导致线粒体功能障碍及线粒体 DNA 释放到胞浆,进而促进NLRP3 炎症小体活化 [17]。Nakahira 等抑制巨噬细胞中的自噬相关蛋白 Atg16L1 或 LC3B 的表达后,发现 NLRP3 炎症小体的活化显著增强 [18]。此外,自噬还可以通过直接降解泛素化的 NLRP3 和 ASC 等 NLRP3 炎症小体主要成分来抑制其活化 [19-20]。此外,一些内源性代谢物质也对 NLRP3 炎症小体有负调控作用,如多巴胺,其膜上受体 DRD1(Dopamine receptor D1)可以活化 cAMP 信号通路促使 cAMP 与 NLRP3 相结合,通过诱导 NLRP3 泛素化及自噬作用来抑制 NLRP3 炎症小体的活化 [19]。β- 羟基丁酸能阻止胞内 K+ 外流,抑制NLRP3 炎症小体的激活 [21]。Mao 等发现 NO 通过增强功能障碍线粒体的清除而抑制 NLRP3 炎症小体的活化 [22]。
4 NLRP3 炎症小体与心血管疾病
4.1NLRP3 炎症小体与动脉粥样硬化
动脉粥样硬化斑块成分如结晶胆固醇和氧化低密度脂蛋白激活NLRP3 炎性体 [23]。Yajima 等人首次使用ASC -/- 小鼠,发现 NLRP3 炎症小体可促进动脉粥样硬化的发生。NLRP3 炎性体负责触发动脉粥样硬化斑块中 IL-1β 的成熟 [24]。ASC 缺乏可降低新生内膜损伤处 IL-1β 和 IL-18 的表达 [25]。彭等人证实 P2X7R 通过促进 NLRP3 炎症小体激活,参与了动脉粥样硬化的进展 [26]。Duewell 等人提出 NLRP3 炎症小体可被胆固醇晶体激活,是动脉粥样硬化形成所必需的 [27]。这些研究均为 NLRP3 炎症小体参与动脉粥样硬化的发生发展提供了新的证据。
4.2NLRP3 炎症小体与高血压
炎症活动在高血压发展中起关键作用,Krishnan 等人观察到高血压患者的血清 IL-1β 水平升高,并大胆猜想,血清IL-1β 升高是否是系统性高血压的标志物或诱导剂 [28]。高盐诱导的炎症和下丘脑室旁核的氧化应激通过交感兴奋作用参与了盐敏感性高血压的发病 [29]。IL-1β 是一种重要促炎细胞因子,中枢抑制 IL-1β 可减少肾素 - 血管紧张素系统激活,减少室旁核中的 ROS 产生,延缓高血压对心血管损伤 [30-31]。在 Dahl 盐敏感性高血压大鼠中,将 IL-1β 抑制剂Gevokizumab 输注入室旁核可抑制交感神经兴奋作用并通过恢复促炎细胞因子与抗炎细胞因子之间的平衡和氧化应激反应来减轻高血压 [32]。
血管紧张素 II (Angiotensin II ,Ang II) 在调节与高血压相关的炎症过程中起关键作用。连续向小鼠体内输注(7 天)Ang II 增强了 NLRP3 炎性小体的激活以及小鼠心脏中IL-1β 和其它促炎性细胞因子的表达,阻断 NLRP3 炎症小体的激活显著减弱了 Ang II 诱导的心脏纤维化而不影响血压, 这表明 NLRP3 炎症小体在 Ang II 诱导的高血压小鼠中独立地对心脏重塑起作用 [33]。
4.3NLRP3 炎症小体与心肌梗塞
心肌梗死(Myocardial infarction,MI)是严重的和(或) 长期的心肌缺血所导致的心肌细胞坏死 , 其主要原因是粥样斑块使冠脉狭窄和(或)不稳定斑块的脱落,而 MI 触发的炎症反应,参与了心肌重塑 [34-35]。研究显示通过干预炎性信号能减少了梗死面积,用抑制 IL-1β 活性的中和抗体和 IL-1 受体拮抗剂 - 阿那白滞素,将明显减轻小鼠急性心肌梗塞后心脏肥大和心肌功能损害,对心肌梗塞小鼠使用抗炎因子IL-10,显著抑制了心肌纤维化,改善心肌重塑并促进了心脏创伤愈合 [36-37]。
5展望
心血管疾病是威胁人类健康的头号杀手,而 NLRP3 炎症小体在高血压、2 型糖尿病等心血管疾病危险因素的发病机制以及动脉粥样硬化和心肌梗塞发展过程中扮演了至关重要的角色,更深层次的探究 NLRP3 炎症小体激活及负调控机制将会为心血管疾病的治疗手段提供更安全、先进、科学的新靶点。然而,目前国内外学者在 NLRP3 炎症小体的探索途中,尚有许多未知的模块,希望广大学者今后能继续对NLRP3 炎症小体保持一颗好奇的心,一生致力于推动人类医疗卫生事业向更安全、更科学的方向前进。
参考文献:
[1]Broz P, Dixit V M. Inflammasomes: mechanism of assembly, regulation and signalling[J]. Nature Reviews Immunology,2016,16(7):407-420.
[2]Bullón P, Cano-García F J, Alcocer-Gómez E, et al. Could NLRP3- Inflammasome Be a Cardiovascular Risk Biomarker in Acute Myocardial Infarction Patients?[J]. Antioxidants & Redox Signaling, 2017, 27(5):269-275.
[3]Hoseini Z, Sepahvand F, Rashidi B, et al. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis[J]. Journal of Cellular Physiolo gy,2017,233(3):2116-2132.
[4]Man S M, Kanneganti T D. Gasdermin D:The long-awaited executioner of pyroptosis[J]. Cell Research,2015,25(11):1183-1184.
[5]Lamkanfi M, Kanneganti T D. Nlrp3: an immune sensor of cellular stress and infection[J]. International Journal of Biochemistry & Cell Biology, 2010, 42(6):792-795.
[6]Lu B, Kwan K, Levine Y A, et al. α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release[J]. Molecular Medicine, 2014, 20(1) :350-358.
[7]刘延刚 , 陈永春 , 宗英 , 等 . NLRP3 炎症小体的活化及调控机制 [J]. 第二军医大学学报 ,2016,(07) :868-872.
[8]Muñozplanillo R, Kuffa P, Martínezcolón G, et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter[J]. Immunity, 2013, 38(6) :1142-1153.
[9]Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor[J]. The EMBO Journal, 2006,25:5071–5082.
[10]Pétrilli V, Papin S, Dostert C, et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration[J]. Cell Death and Differentiation, 2007, 14(9) :1583-1589.
[11]Murakami T, Ockinger J, Yu J, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome[J]. PNAS,2012,109(28):11282-11287.
[12]Torreminguela C D, Castillo P M D, Pelegrín P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases[J]. Frontiers in Immunology, 2017,43(8).
[13]Orlowski G M, Colbert J D, Sharma S, et al. Multiple cathepsins promote pro-IL-1 beta synthesis and NLRP3-mediated IL-1 beta activation (vol 195, pg 1685, 2015)[J]. Journal of Immunology, 2016, 196(1) :503-503.
[14]Zhou R, Yazdi A S, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation[J]. Nature, 2011, 469(7329) :221-225.
[15]Shenker B J, Ojcius D M, Walker L P, et al. Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin Activates the NLRP3 Inflammasome in Human Macrophages, Leading to the Release of Proinflammatory Cytokines[J]. Infection & Immunity,2015,83(4):1487-1496.
[16]Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production[J]. Nature, 2008, 456(7219):264-268.
[17]Lupfer C, Thomas P G, Anand P K, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection[J]. Nature Immunology, 2013, 14(5) :480-488.
[18]Nakahira K, Haspel J A, Rathinam V A, et al. Autophagy Proteins Regulate Innate Immune Response By Inhibiting NALP3 Inflammasome-Mediated Mitochondrial DAN Release[J]. American Thoracic Society International Conference, 2011, 12 (3):222-230.
[19]Yan Y, Jiang W, Liu L, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome[J]. Cell,2015,160(1-2):62-73.
[20]Shi C S, Shenderov K, Huang N N, et al. Activation of autophagy by inflammatory signals limits IL-1[beta] production by targeting ubiquitinated inflammasomes for destruction[J]. Nature Immunology,2012,13(3):255-263.
[21]Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease[J]. Nature Medicine,2015,21(3):263-269.
[22]Mao K, Chen S, Chen M, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock[J]. Cell Research,2013,23(2):201-212.
[23]Hoseini Z, Sepahvand F, Rashidi B, et al. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis[J]. Journal of Cellular Physiolo gy,2018,233(3):2116-2132.
[24]Cochain C, Zernecke A. Protective and pathogenic roles of CD8(+) T cells in atherosclerosis[J]. Basic Research in Cardiology,2016,111(6):71.
[25]Yajima N, Takahashi M, Morimoto H, et al. Critical role of bone marrow apoptosis-associated speck-like protein, an inflammasome adaptor molecule, in neointimal formation after vascular injury in mice[J]. Circulation, 2008, 117(24) :3079-3087.
Peng K, Liu L, Wei D, et al. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation[J].
International Journal of Molecular Medicine, 2015, 35(5) :1179-1188.
[27]Duewell P, Kono H, Rayner K J, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals[J]. Nature, 2010, 464(7293) :1357-1361.
[28]Krishnan SM, Sobey CG, Latz E, et al. IL-1β and IL-18: Inflammatory markers or mediators of hypertension?[J]. British Journal of Pharmacology, 2014, 171(24) :5589-5602.
[29]Rust P, Ekmekcioglu C. Impact of Salt Intake on the Pathogenesis and Treatment of Hypertension[J]. Advances in Experimental Medicine & Biology,2016,956:61-84.
[30]Fujita T. Mechanism of salt-sensitive hypertension: focus on adrenal and sympathetic nervous systems[J]. Journal of the American Society of Nephrology Jasn, 2014, 25(6) :1148-1155.
[31]Su Q, Qin D N, Wang F X, et al. Inhibition of reactive oxygen species in hypothalamic paraventricular nucleus attenuates the renin-angiotensin system and proinflammatory cytokines in hypertension[J]. Toxicology & Applied Ph armacology,2014,276(2):115-120.
[32]Qi J, Zhao X F, Yu X J, et al. Targeting Interleukin-1 beta, to Suppress Sympathoexcitation in Hypothalamic Paraventricular Nucleus in Dahl Salt- Sensitive Hypertensive Rats[J]. Cardiovascular Toxicology,2016,16(3):298-306.
[33]Gan W, Ren J, Li T, et al. The SGK1 inhibitor EMD638683, prevents Angiotensin II-induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation[J]. Biochimica Et Biophysica Acta,2017,1864(1):1-10.
[34]Frangogiannis N G. The inflammatory response in myocardial injury, repair and remodeling[J]. Nature Reviews Cardiology,2014,11(5):255-265.
[35]Takahashi M. Role of the SDF-1/CXCR4 system in myocardial infarction[J]. Circulation Journal Official Journal of the Japanese Circulation Society,2010,74(3):418-423.
[36]Jung M, Ma Y, Iyer R P, et al. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation[J]. Basic Research in Cardiology,2017,112(3):33.
[37]Rienks M, Carai P, Bitsch N, et al. Sema3A promotes the resolution of cardiac inflammation after myocardial infarction[J]. Basic Research in Cardiology,2017,112(4):42.
《NLRP3炎症小体及其与心血管疾病的关系论文》附论文PDF版下载:
http://www.lunwensci.com/uploadfile/2018/1030/20181030024211702.pdf
关注SCI论文创作发表,寻求SCI论文修改润色、SCI论文代发表等服务支撑,请锁定SCI论文网!
文章出自SCI论文网转载请注明出处:https://www.lunwensci.com/yixuelunwen/1364.html