SCI论文(www.lunwensci.com):
摘要:目的本文拟对原发性肌张力障碍基因芯片数据进行生物信息学分析。方法从GEO数据库获取原发性肌张力障碍的标志物芯片表达的差异基因,应用生物信息学方法对差异基因进行功能注释和通路分析,string-db数据库进行蛋白相互作用分析。结果经过数据分析,这些在差异性表达的基因被富集到不同的生物学过程,主要富集在serine-type肽酶过程和胰岛素受体复合这两种过程。结论原发性肌张力障碍可能存在较为复杂的生物过程。
关键词:原发性肌张力障碍;基因芯片;生物信息学;生物标志物
本文引用格式:戴冠东,王凯,罗丽丹.通过生物信息学方法探索家族性成人原发性肌张力障碍中相关基因表达[J].世界最新医学信息文摘,2019,19(92):128-130.
0引言
原发性肌张力障碍(primary torsion dystonia,PTD)是一种主动肌与拮抗肌收缩不协调或过度收缩引起的以异常姿势和动作为特征的椎体外系疾病。PTD常累及面部,喉咙及颈部,往往持续保持局灶性或阶段性。以成人起病的原发性局限性肌张力障碍中,15%-30%可发展至肢体的其他部位[1]。有学者将原发性肌张力障碍定义为一种由病因-病理和临床标准,多为“特发性”震颤或“特发性”帕金森疾病[2]。排除标准,包括病理、病史和检查这些发现对于将原发性肌张力障碍与其他肌张力障碍区分开来一直是至关重要的。奥本海姆于1911年描述了该疾病的临床表现:肌肉张力的间歇性变化、有节奏的抽搐、扭转挛缩和姿势[3-4]。同时,他指出肌张力障碍是一个独立的病态实体。当前较多文献指出,该病是与很多神经退行性变和神经递质代谢紊乱相关的症状群。以较多散发,少数有家族史,该病的发生与遗传和环境有关[5-7]。有证据表明,不光是家族性肌张力障碍具有一定遗传基础,极可能很多明显散发性的病例也有遗传基础[8]。随着越来越多致病基因或易感基因的定位、发病机制的逐步阐明,临床医师势必能够利用这些信息更好地发现每种遗传类型的临床特征,从而进行更有效的治疗。
随着生物信息学发展,本研究通过检索全球性公共数据库来寻找与家族性成人原发性肌张力障碍相关基因芯片,并应用生物信息学技术来探索较为显著的差异基因,并绘制与之相关的调控网络,研究多个基因之间的相互联系。
1材料与方法
1.1数据集的获取和资料分析
以“Primary dystonia”为关键词对美国国立生物中心的GEO数据库(http:www.ncbi.nlm.nih.gov/geo)进行检索,获得美国田纳西大学健康科学中心公布的相关基因芯片数据集GSE43771。相关研究芯片平台为GPL10558Illumina HumanHT-12 V4.0 expression beadchip。该实验以人类外周血为研究对象,纳入对象为成人原发性肌张力障碍家族,共8个样品,4名携带者,4名非携带者。
1.2家族性成人原发性肌张力障碍差异表达基因的分析
相关基因差异表达分析,采用R语言程序包,根据统计学方法对基因进行方差分析及相关的t检验,来鉴定差异基因。本研究中将4名gα(olf)家族性成人原发性肌张力障碍携带者作为实验组,4名gα(olf)家族性成人原发性肌张力障碍非携带者样本作为对照组,分析两组样本差异表达的基因。差异倍数阈值设置为log≥1.5且P<0.01。
1.3相关差异基因阈值设定
筛选出来的差异基因通过DAVID数据库中进行相关KEGG通路富集,P值及错误发生率相关阈值设定(false discovery rate,FDR)均≤0.01,符合该阈值标准则视为具有统计学意义的生物通路。String相关数据程序库进行相应的蛋白间的交互作用分析。
2结果
经预处理后可知,每个样本的芯片数据值分布较好(图1)。根据当前统计学中的贝叶斯分析法处理后,经排除未检索到的基因,共获得336个基因的差异表达≥1.5倍,包括215个上调显著基因和121个下调基因,最终保留上调和下调各10个显著基因,如表1所示。
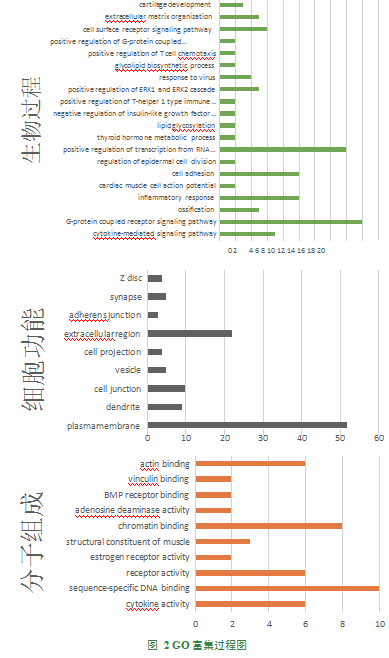
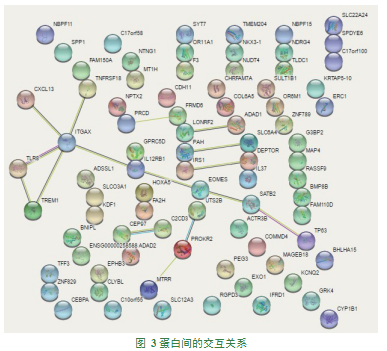
通过基因差异表达分析所筛选出的差异个基因进行富集其中,较为显著的生物过程为RNA聚合酶II启动子转录的正调控,较为显著的细胞功能为细胞质膜,较为显著的分子功能为序列特异性DNA结合蛋白,见图2;相关基因间的交互关系如图3;较为显著的离子通路为粘附连接通路。
3讨论
原发性肌张力障碍通常为单基因遗传病,以常染色体显性遗传伴不同外显率为主,无明确的神经病理学改变。随着遗传学的快速发展,迄今为止共发现20余种基因与原发性肌张力障碍相关。2012年Tania等[8-11]通过对两个原发性扭转型肌张力障碍家系进行全外显子测序,将其致病基因定位到GNAL基因。GNAL位于18p11.22-p11.21,共有,12个外显子,编码兴奋性α亚单位一Gα(olf)。Gα(olf)有3个异构体:异构体1(NM_182978.3)是GNAL基因编码的最长蛋白,异构体2(NM_001142339.2)—是最主要的蛋白,两者同时存在于大脑和白细胞中,在大脑皮层和纹状体含量最多[14],异构体3在脑中不存在。脑中最主要的兴奋性G蛋白亚单位是Gαs,但在纹状体棘状神经元(striatal medium spiny neurons,MSNs)中,Gα(olf)取代Gα。Gα(olf)与直接通路中多巴胺1型受体(D1Rs)及间接通路中腺苷A2A受体(A2ARs)结合,激活5型腺苷酸环化酶[11]。GNAL基因突变导致Gα(olf)亚单位的合成受阻,出现功能障碍。GNAL基因突变形式多样,包括无义突变,移码突变,错义突变、框内缺失和剪接异常等。Vemula等在760名肌张力障碍患者中发现3个突变位点[11],MIAO在名中国东北部患者中发现2个突变[12],Kumar对318名患者中发现2个突变[13]总体来讲,目前国际上对GNAL基因尚在初步研宄阶段,国内尚缺乏大样本人群研究。
在肌张力障碍领域,全基因组关联研究(genome-wide association studies,GWAS)第一次发现阵发性运动障碍(paroxysmal kinesigenic dyskinesia,PKD)DYT10位点,这是一种罕见的偶发性肌张力障碍综合征,以突然随意运动引发重复和短暂的不自主发作作为特点。其重叠的表型,包括小二发作性舞蹈手足徐动症(infantile convulsions and paroxysmal choredo athetosis,ICCA)和良性家族性小二癫痫综合征(benign familial infantile seizures syndrome,BFIS)PKD、ICCA和其他表型相关基因定位在16号染色体上,最近的研究运用全外显子测序(whole exome sequencing,WES)方法证实Proline-rich transmembrane protein2(PRRRT2)基因的突变为PKD和ICCA的主要原因[14],WES是指利用序列捕获技术将全基因组外显子区域DNA捕获并富集后进行高通量测序的基因组分析方法。随后,同样的WES方法也陈宫用于识别4个迟发性PTD的新诱发基(TUBB4a、CIZ1、ANO3、GNAL)[15]。这证明相同基因的突变或导致异构表型[15]。全基因组关联研究旨在通过大规模、发杂疾病人权的研究识别多态变量作为复杂疾病的风险因素。在大群患者和对照组中,不计其数的常见变异(次要等位基因频率大于5%)可同时被基因分型,需要严谨的统计分析,从假阳性结果中分辨真实的结果。这种方法已基本适用于PTD,尽管显著阳性结果的数量出人意料的低,但也发现了几个藏匿于疾病中关联变体的基因,例如SNCA、MAPT和LRRK2。到目前为止,没有关于PTD的GWAS的报道,但是该技术在此领域的潜在贡献确实显而易见。大多数成人发病型原发性肌张力障碍的病因仍然不明确。该病具有临床异质性与遗传异质性,随着分子遗传学的迅速发展,很多肌张力障碍的基因位点被相继发现,目前报道有25种肌张力障碍的基因位点(DYT1~DYT25)[16-17]。
在本研究中,从新的角度通过用生物信息学方法对家系内的发病人群和非发病人群,进行对比,结果发现两组人群中较为显著的基因为BNIPL、MIR412、TMEM204,这三种基因在水肿和炎性疾病中发挥重要作用,而在蛋白间的交互作用中较为显著的是ITGAX,筛选出的较为显著的生物过程为RNA聚合酶II启动子转录的正调控,较为显著的细胞功能为细胞质膜,较为显著的分子功能为序列特异性DNA结合蛋白,相关基因间的交互关系,较为显著的离子通路为粘附连接通路。由此,可见该疾病的发生和发展,与基因的调控有一定的关联,可能存在新的路径或通路。同时也从一定程度上验证了,与全基因组测序相同的结论,该病的发病与基因调控存在一定的关联性。
目前对家族性的原发性肌张力障碍疾病的研究较少,该病主要靠药物和立体定向手术治疗,但这些治疗只是对症治疗,并有很多局限性,并且肌张力障碍的发病机制仍未完全清楚,也无有效的治疗方法;因此还需要进一步对家族性的疾病基因进行的新位点的筛查,对发现新的相关基因及相关蛋白进行深入的研究,提供一定的理论基础。
参考文献
[1]Dewey FE,Chen R,Cordero SP,et al.Phased whole-genome genetic risk in a family quartet using a major allele reference sequence[J].PLoS Genet,2011,7(9):e1002280.
[2]Kumar KR,Lohmann K,Klein C.Genetics of Parkinson disease and other movement disorders[J].Curr Opinion Neurol,2012,25(4):366-474.
[3]Slingsby,C.and G.J.Wistow,Functions of crystallins in and out of lens:Roles in elongated and post-mitotic cells[J].Prog Biophys Mol Biol,2014,115(1):52-6.
[4]Parker,M.,et al.Suppression of neovascularisation of donor corneas by transduction with Equine infectious anemia virus-based lentiviral vectors expressing endostatin and angiostatin[J].Hum Gene Ther,2014,25(5):408-418.
[5]Ozelius LJ,Bressman SB.Genetic and clinical features of primary torsion dystonia[J].Neurobiol Dis,2011,42(2):127-135.
[6]Bressman SB,Greene PE.Dystonia[J].Curr Treat Options Neurol,2000,2(3):275-285.
[7]Lee HY,Huang Y,Bruneau N,et al.Mutations in the gene PRRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions[J].Cell Rep,2012,1(1):2-12.
[8]Fuchs T,Saunders-pullman R,Masuho I,et al.Mutations in GNAL cause primary torsion dystonia[J].Nat genet,2013,45(1):88-92.
[9]Vemula SR,Puschmann A,Xiao J,et al.Role of Galpha(olf)in familial and sporadic adult-onset primary dystonia[J].Hum Mol Genet,2013,22(12):2510-9.
[10]Kull B,Svenningsson P,Fredholm BB.Adenosine a(2A)receptors are colocalized with and aceivate g(olf)in rat striatum[J].Mol Pharmacol,2000,58(4):777-7.
[11]Fuchs T,Saunders-pullman R,Masuho I,et al.Mutations in GNAL cause primary torsion dystonia[J].Nat genet,2013,45(1):88-92.
[12]Vemula SR,Puschmann A,Xiao J,et al.Role of Galpha(olf)in familial and sporadic adult-onset primary dystonia[J].Hum Mol Genet,2013,22(12):2510-9.
[13]Kull B,Svenningsson P,Fredholm BB.Adenosine a(2A)receptors are colocalized with and aceivate g(olf)in rat striatum[J].Mol Pharmacol,2000,58(4):777-7.
[14]Miao J,Wan XH,Sun Y,et al.Mutation sceeeing of GNAL gene in patients with primary dystonia from Northeast China[J].Parkinsonism Relat Disord,2013,19(10):910-2.
[15]Kumar KR,Lohmann K,Masuho I,et al.Mutations in GNAL:A Novel Cause of Craniocervical Dystonia[J].JAMA Neutol,2014.
[16]Fuchs T,Saunders-Pullman R,Masuho I,et al.Mutations in GNAL cause primary torsion dystonia[J].Nat Genet,2013,45(1):88-92.
[17]Schmidt A,Kumar KR,Redyk K,et al.Two Faces of the Same Coin:Benign Familial Infantile Seizures and Paroxysmal Kinesigenic Dyskinesia Caused by PRRT2 Mutations[J].Archives of Neurology,2012,69(5).
关注SCI论文创作发表,寻求SCI论文修改润色、SCI论文代发表等服务支撑,请锁定SCI论文网! 文章出自SCI论文网转载请注明出处:https://www.lunwensci.com/yixuelunwen/24749.html